A list of weird gene annotations or things that break bioinformatics assumptions
See also https://github.com/cmdcolin/oddbiology/ for more weird bio
Evidence given for a 1bp length exon in Arabidopsis and different splicing models are discussed
http://www.nature.com/articles/srep18087
Another 1bp exon is discussed here https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0177959
Microexons in general are an interesting topic and are "involved in important biological processes in brain development and human cancers" (ref https://www.cell.com/molecular-therapy-family/nucleic-acids/fulltext/S2162-2531(23)00013-6) yet are commonly misannotated (e.g. in plants https://www.nature.com/articles/s41467-022-28449-8)
See also cryptic splice sites, cryptic exons, poison exons
The phenomenon of recursive splicing can remove sequences progressively inside an intron, so there can exist "0bp exons" that are just the splice-site sequences pasted together.
"To identify potential zero nucleotide exon-type ratchet points, we parsed the RNA-Seq alignments to identify novel splice junctions where the reads mapped to an annotated 5' splice site and an unannotated 3' splice site, and the genomic sequence at the 3' splice site junction was AG/GT"
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4529404/
It was found that aberrant recursive splicing could potentially contribute to disease https://www.biorxiv.org/content/10.1101/2025.08.14.666599v1?med=mas
Satellite DNA study uncovers megabase scale introns https://www.biorxiv.org/content/early/2018/12/11/493254
An example in this paper kl-3 spans 4.3 million bp
In human, an example is Dystrophin. "Dystrophin is coded for by the DMD gene – the largest known human gene, covering 2.4 megabases (0.08% of the human genome) at locus Xp21. The primary transcript in muscle measures about 2,100 kilobases and takes 16 hours to transcribe; the mature mRNA measures 14.0 kilobases" https://en.wikipedia.org/wiki/Dystrophin
Note: these large introns require very large amounts of DNA to be transcribed into RNA, before just removing most of the transcribed RNA via intron splicing, which is sort of "wasteful" on a molecular level. The 16-hour transcription time for dystrophin means that rapidly dividing cells cannot finish transcribing it before the next cell division interrupts the process https://pmc.ncbi.nlm.nih.gov/articles/PMC2754300/
In human, the TTN (titin) gene has ~364 exons, which is almost double the next most NEB (nebulin) at ~184 exons
https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=7273
"A 2015 study suggests that the shortest known metazoan intron length is 30 base pairs (bp) belonging to the human MST1L gene (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4675715/). The shortest known introns belong to the heterotrich ciliates, such as Stentor coeruleus, in which most (> 95%) introns are 15 or 16 bp long (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5659724/)" https://en.wikipedia.org/wiki/Intron#Distribution
A novel splicing factor may be involved in small introns https://www.news-medical.net/news/20240215/Novel-splicing-mechanism-for-short-introns-discovered.aspx
An alga described in 2024 encodes a protein PKZILLA-1 that has a mass of 4.7 megadaltons and contains 140 enzyme domains https://cen.acs.org/biological-chemistry/PKZILLA-proteins-smash-protein-size/102/web/2024/08
In human the TTN gene encodes the titin protein (in muscle) at almost 4 megadaltons

The DMD gene above, despite being the largest known human gene (2.4 Mb), encodes a ~427 kDa protein — large, but nowhere near megadalton scale. A shorter isoform Dp71 (~71 kDa) is expressed in non-muscle tissues. https://pmc.ncbi.nlm.nih.gov/articles/PMC49288/
Inverted Alu pairs flanking an exon can fold into an RNA stem-loop hairpin that physically loops the exon out, causing skipping through RNA secondary structure alone rather than splicing factors. ~707 human exons are affected, including TBXT (linked to tail loss in hominoids).
https://academic.oup.com/nar/article/54/6/gkag196/8539533
The process of "backsplicing" circularizes RNAs. There can be alternative backsplicing too

Figure from Dawoud et al. https://doi.org/10.1016/j.ncrna.2022.09.011
See https://academic.oup.com/nar/article/48/4/1779/5715065
"Dscam has 24 exons; exon 4 has 12 variants, exon 6 has 48 variants, exon 9 has 33 variants, and exon 17 has two variants. The combination of exons 4, 6, and 9 leads to 19,008 possible isoforms with different extracellular domains (due to differences in Ig2, Ig3 and Ig4). With two different transmembrane domains from exon 17, the total possible protein products could reach 38,016 isoforms"
Ref https://en.wikipedia.org/wiki/DSCAM https://www.wikigenes.org/e/gene/e/35652.html
Ref https://en.wikipedia.org/wiki/Translational_frameshift
https://www.sciencedirect.com/topics/neuroscience/ribosomal-frameshifting
SARS-CoV-2 uses ribosomal frameshifting and this video shows a 3D animation of the process, showing a 'pseudoknot' in the RNA contributes to it https://www.youtube.com/watch?v=gLcueW61QMU
Another lecture explaining frameshift in viruses https://youtu.be/b5BX5A3dGUQ?t=2980
In retroviruses like HIV, the gag and pol genes overlap in different reading frames. A -1 ribosomal frameshift at a "slippery sequence" between them produces the Gag-Pol fusion polyprotein at ~5% efficiency, while the other 95% of ribosomes terminate at the gag stop codon and produce only Gag. This ratio is critical — altering it is lethal to the virus. The Gag-Pol polyprotein is then cleaved by the viral protease (which is itself part of the polyprotein) to produce reverse transcriptase, integrase, and protease.
https://en.wikipedia.org/wiki/Gag-pol https://en.wikipedia.org/wiki/Pol_(HIV)

Figure from Atkins et al. (2016) showing the ribosome encountering a slippery sequence (X XXY YYZ) and downstream RNA stimulatory element (pseudoknot) that together promote -1 programmed ribosomal frameshifting https://pmc.ncbi.nlm.nih.gov/articles/PMC7618472/
"Ribosome hopping involves ribosomes skipping over large portions of an mRNA without translating them" Ref https://pubmed.ncbi.nlm.nih.gov/24711422/
The classic example is bacteriophage T4 gene 60, where the ribosome bypasses a 50-nucleotide coding gap — about half of ribosomes successfully make the hop https://pmc.ncbi.nlm.nih.gov/articles/PMC107096/
An IRES allows ribosomes to initiate translation at an internal position on an mRNA without scanning from the 5' cap. This enables cap-independent translation and is used by many viruses to hijack host ribosomes. Some cellular mRNAs also contain IRES elements, allowing them to be translated under stress conditions when cap-dependent translation is shut down.
https://en.wikipedia.org/wiki/Internal_ribosome_entry_site
"Stop codon suppression or translational readthrough occurs when in translation a stop codon is interpreted as a sense codon, that is, when a (standard) amino acid is 'encoded' by the stop codon. Mutated tRNAs can be the cause of readthrough, but also certain nucleotide motifs close to the stop codon. Translational readthrough is very common in viruses and bacteria, and has also been found as a gene regulatory principle in humans, yeasts, bacteria and drosophila.[28][29] This kind of endogenous translational readthrough constitutes a variation of the genetic code, because a stop codon codes for an amino acid. In the case of human malate dehydrogenase, the stop codon is read through with a frequency of about 4%.[30] The amino acid inserted at the stop codon depends on the identity of the stop codon itself: Gln, Tyr, and Lys have been found for the UAA and UAG codons, while Cys, Trp, and Arg for the UGA codon have been identified by mass spectrometry.[31] Extent of readthrough in mammals have widely variable extents, and can broadly diversify the proteome and affect cancer progression.[32] "
https://en.wikipedia.org/wiki/Stop_codon#Translational_readthrough
The amino acid Selenocysteine is coded for by an "opal" (UGA) stop codon (https://en.wikipedia.org/wiki/Selenocysteine)
Is present in all domains of life including humans
As of 2021, 136 human proteins (in 37 families) are known to contain selenocysteine
A stem-loop structure in the mRNA called a SECIS element (https://en.wikipedia.org/wiki/SECIS_element) signals the ribosome to read UGA as selenocysteine instead of stop. The resulting products are called selenoproteins.
Pyrrolysine also is coded for by the "amber" (UAG) stop codon (https://en.wikipedia.org/wiki/Pyrrolysine), not present in humans
"It is encoded in mRNA by the UAG codon, which in most organisms is the 'amber' stop codon. This requires only the presence of the pylT gene, which encodes an unusual transfer RNA (tRNA) with a CUA anticodon, and the pylS gene, which encodes a class II aminoacyl-tRNA synthetase that charges the pylT-derived tRNA with pyrrolysine. "
There are several other stop codon modifications described here https://www.nature.com/articles/nrg3963
In mammalian apolipoprotein B, RNA editing introduces a premature stop codon rather than removing one. The default unedited mRNA encodes the full-length B100 isoform (550 kDa). In the intestine, C-to-U editing at nucleotide 6666 converts a glutamine codon (CAA) into a stop codon (UAA), producing the truncated B48 isoform (265 kDa). This is one of the best-characterized examples of RNA editing altering protein output.
https://pubmed.ncbi.nlm.nih.gov/8409768/
"Flexibility in the nuclear genetic code has been demonstrated in ciliates that reassign standard stop codons to amino acids...Surprisingly, in two of these species, we find efficient translation of all 64 codons as standard amino acids and recognition of either one or all three stop codons"
Translation termination is therefore "context dependent" — determined by surrounding sequence rather than just the 3-letter codon https://pubmed.ncbi.nlm.nih.gov/27426948/
See also how Ciliates perform programmed DNA elimination (see that section) and have some of the smallest known introns (see small introns section)
See also this Ensembl blog on annotating readthrough transcription which joins multiple genes http://www.ensembl.info/2019/02/11/annotating-readthrough-transcription-in-ensembl/
RNA-seq often makes extremely compelling cases for two-or-more different genes to be conjoined by splicing
Some algorithms e.g. mikado https://academic.oup.com/gigascience/article/7/8/giy093/5057872 try to avoid this calling it artifactual fusion/chimera that can be due to some tandem duplication but it does seem to be very prevalent in real data sets
The standard splice site is GT on the 5' end (donor) and AG on the 3' end (acceptor), processed by the major (U2-type) spliceosome. Non-GT-AG sites exist: GC-AG is the most common variant (~1% of introns) and is still handled by the major spliceosome. A separate minor (U12-type) spliceosome handles a distinct class of introns, originally identified by AT-AC termini but now known to also include GT-AG introns with different internal sequences.
https://en.wikipedia.org/wiki/Minor_spliceosome
Cryptic splice sites are sequences that resemble splice sites but are normally not used. They can be activated by mutations that disrupt normal splice sites, or by changes in splicing factor expression. Deep intronic variants are a major source, accounting for a large fraction of splice-disrupting events in genetic disease.
Review article https://academic.oup.com/nar/article/39/14/5837/1382796
The snaptron project from Ben Langmead analyzed huge amounts of RNA-seq public data and found many types of these cryptic splicing http://snaptron.cs.jhu.edu/
SpliceVault and similar tools re-analyze large public RNA-seq datasets to empirically predict cryptic splice site activation https://www.nature.com/articles/s41588-022-01293-8
See also cryptic exons, poison exons
Cryptic exons are intronic sequences that get erroneously included as exons when cryptic splice sites flanking them become activated. They usually introduce a premature termination codon (PTC) that triggers nonsense-mediated decay (NMD), effectively reducing gene expression. This makes them hard to detect since the aberrant transcripts are often degraded.
A prominent example involves TDP-43 in ALS/FTD: TDP-43 normally represses inclusion of nonconserved cryptic exons, and when TDP-43 is depleted from the nucleus (a hallmark of ALS/FTD pathology), cryptic exons are included in key neuronal mRNAs like STMN2 and UNC13A https://www.science.org/doi/10.1126/science.aab0983 https://www.nature.com/articles/s41586-022-04424-7
Review: https://link.springer.com/article/10.1186/s13024-023-00608-5
Poison exons are conserved alternative exons that contain a premature termination codon (PTC). When included via alternative splicing, the PTC triggers NMD, degrading the transcript. Unlike cryptic exons which are aberrantly activated, poison exons are a deliberate gene regulation mechanism: cells fine-tune protein levels by adjusting the ratio of productive (poison-exon-skipped) vs. NMD-targeted (poison-exon-included) transcripts.
Splicing factors like SR proteins and hnRNPs autoregulate their own levels via poison exon inclusion. Therapeutically, ASOs that block poison exon inclusion can upregulate expression of disease-relevant genes.
Review https://www.sciencedirect.com/science/article/pii/S0165614725000021
Poison exons in neurodevelopment and disease https://pmc.ncbi.nlm.nih.gov/articles/PMC8042789/
Exitrons (exonic introns) are internal regions within protein-coding exons that can be spliced out like introns. When an exitron is retained it contributes to the protein sequence; when spliced out it causes an in-frame deletion or frameshift.
Exitron splicing is dramatically elevated in cancer: one study found 63% of human coding genes are affected in tumors vs. 17% in normal tissue, and they can generate tumor-specific neoepitopes https://www.cell.com/molecular-cell/fulltext/S1097-2765(21)00223-9
Original paper defining exitrons https://genome.cshlp.org/content/25/7/995.full
NAGNAG, GYNGYN, repeats of the splicing signal cause modified splicing behavior
"Another mechanism introducing small variations to protein isoforms is wobble splicing. Here, a GYN repeat at the donor splice site (5’ splice site; Y stands for C or T and N stands for A, C, G, or T) or an NAG repeat at the acceptor splice site (3’ splice site) leads to subtle length variations in the spliced transcripts and finally to alternative isoforms differing in few amino acids." ref https://onlinelibrary.wiley.com/doi/full/10.1002/bies.201900066?af=R
Intron retention (IR) is a phenomenon where intron sequence is preserved, or doesn't get spliced out, in mature RNA
It can occur in both abnormal and normal biological conditions. Transcript with IR often undergo nonsense-mediated decay.
https://en.wikipedia.org/wiki/Alternative_splicing
Review: https://pmc.ncbi.nlm.nih.gov/articles/PMC12385487/
Normally RNA is spliced by the spliceosome, a ribonucleoprotein complex where the catalysis is actually performed by its RNA components (snRNAs). There is also self-splicing RNA where the RNA catalyzes its own splicing without the spliceosome
Group I introns use a free guanosine as a cofactor to catalyze their own excision https://en.wikipedia.org/wiki/Group_I_catalytic_intron
Group II introns use a different mechanism (lariat formation, similar to spliceosomal introns) and are thought to be evolutionary ancestors of the spliceosome https://en.wikipedia.org/wiki/Group_II_intron
There are some small intron types called "bulge-helix-bulge" in archaea (and other organisms)

From https://www.embopress.org/doi/full/10.1038/embor.2008.101
The figure above shows that the orange part is excised as an intron for the tRNA
A twintron is an intron-within-an-intron. The internal intron must be spliced first before the outer one can be recognized and spliced (if the internal one does not need to be spliced first, it is simply called a nested intron).
See https://en.wikipedia.org/wiki/Twintron

Figure from https://doi.org/10.1080/15476286.2015.1103427 showing twintron conformations with a) spliceosomal introns b) group I/II self-splicing introns and c) tRNA/bulge helix bulge type introns
Introns were first discovered in adenoviruses (1977), not eukaryotes — a finding that won Roberts and Sharp the Nobel Prize
https://en.wikipedia.org/wiki/Intron#Discovery_and_etymology (see also https://www.proquest.com/docview/303935681/)
Pieces of the mitochondrial genome can be inserted into nuclear chromosomes in eukaryotes. The human genome contains over 700 NUMTs. They confound mitochondrial studies because PCR primers for mtDNA can accidentally amplify NUMTs, and new insertions continue to occur — some are pathogenic.
https://en.wikipedia.org/wiki/Nuclear_mitochondrial_DNA_segment https://pmc.ncbi.nlm.nih.gov/articles/PMC3228813/
Many eukaryotes use the "standard genetic code" for translating codons to amino acids, but alternative codes are common across the tree of life — especially in mitochondria, ciliates, and mycoplasma. The NCBI genetic code table lists these variants
https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi#SG31
One article explains how alternative genetic codes work https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6207430/
In vertebrate mitochondria, UGA codes for tryptophan instead of stop, and AGA/AGG code for stop instead of arginine — a direct reversal of the standard code https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi#SG2
The number of known alternative codes keeps growing — NCBI is currently up to table 33, and computational screening of 250,000+ genomes (including metagenome-assembled genomes) has found additional codes not yet officially adopted https://elifesciences.org/articles/71402
The 5' and 3' UTR (untranslated region) are parts of the mature mRNA that are not translated into protein — the 5' UTR is before the start codon, the 3' UTR is after the stop codon. Unlike introns, UTRs are retained in the final mRNA. They are often targets of miRNA binding which can lead to degradation.
This blog post by Ensembl shows how they annotate UTR and a 19kb 3' UTR in Grin2b http://www.ensembl.info/2018/08/17/ensembl-insights-how-are-utrs-annotated/
Polyadenylation is the addition of ~200 adenines to the 3' end of an mRNA. A polyadenylation signal (typically AAUAAA) in the pre-mRNA is recognized by the cleavage and polyadenylation complex, which cleaves the RNA and adds the poly-A tail. The tail is not encoded in the genome. It protects the mRNA from degradation and aids nuclear export and translation. https://en.wikipedia.org/wiki/Polyadenylation
A survey of poly-A using Oxford Nanopore found a transcript isoform with a 450bp poly-A tail (ENST00000581230), with intron retention being a possible correlate of longer poly-A tails https://www.biorxiv.org/content/early/2018/11/09/459529.article-info
Polyadenylation normally happens at the end of a transcript, after the last exon. "Intronic polyadenylation" (IpA) is the confusingly-named phenomenon where a poly-A signal hiding inside an intron gets used instead. When this happens, the cell treats that intronic position as "the end" of the transcript — everything downstream (including remaining exons) is lost. The result is a shorter mRNA that may encode a truncated protein or no functional protein at all. The name is confusing because "intronic" suggests something happening inside an intron that gets spliced away, but here the intron is where transcription effectively stops. The classic example is immunoglobulin M (IgM): naive B cells use a downstream poly-A site to produce membrane-bound IgM, but upon activation, an intronic poly-A site is used instead, producing the shorter secreted form of IgM. This switch is a key part of the immune response (https://pmc.ncbi.nlm.nih.gov/articles/PMC9715272/, https://pubmed.ncbi.nlm.nih.gov/9885564/).

Figure showing "intronic polyadenylation" (IpA) creating a different isoform from https://www.nature.com/articles/s41467-018-04112-z
In mammalian mitochondria, some mRNAs are polyadenylated immediately after a genomically encoded U — the poly-A tail supplies the two A's needed to complete a UAA stop codon. Without polyadenylation, these transcripts would lack a stop codon entirely (noted in https://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi#SG2).
Circularized chromosomes should be unsurprising to anyone working with plasmids and many prokaryotic genomes but for gene annotation formats which use linear coordinates, representing anything wrapping around the origin is challenging.
While mitochondrial genomes are often assumed to be circular, some eukaryotes have linear mitochondrial DNA — including certain yeasts (Candida) and some plants and protists https://www.sciencedirect.com/science/article/abs/pii/S0168952503003044
Many genomic viewers do not do this well. For GFF format this is done by making the end go past the end of the genome. Below, the genome is 6407 bp in length, but the CDS feature extends past this and sets Is_circular=true
##gff-version 3.2.1
# organism Enterobacteria phage f1
# Note Bacteriophage f1, complete genome.
J02448 GenBank region 1 6407 . + . ID=J02448;Name=J02448;Is_circular=true;
J02448 GenBank CDS 6006 7238 . + 0 ID=geneII;Name=II;Note=protein II;
GFF3 specification: https://github.com/the-sequence-ontology/specifications/blob/master/gff3.md
The replication of the 2 micron plasmid found in Saccharomyces cerevisiae relies on a programmed DNA rearrangement; in any population of cells two different states of the 2 micron plasmid can be expected and these will interconvert in later generations. Reference: https://pubmed.ncbi.nlm.nih.gov/23541845/
It is possible for gene sequences to overlap, on different strands (sense-antisense) or same strand, possibly in alternate coding frames
https://en.wikipedia.org/wiki/Overlapping_gene
Some articles
- The novel EHEC gene asa overlaps the TEGT transporter gene in antisense and is regulated by NaCl and growth phase https://www.ncbi.nlm.nih.gov/m/pubmed/30552341/
- Overlapping genes in natural and engineered genomes https://www.nature.com/articles/s41576-021-00417-w
- Uncovering de novo gene birth in yeast using deep transcriptomics https://www.nature.com/articles/s41467-021-20911-3
Hepatitis B is an extreme example of overlapping genes — its ~3.2 kb genome packs 4 overlapping reading frames, with every nucleotide belonging to at least one gene https://pmc.ncbi.nlm.nih.gov/articles/PMC3126273/

Figure from https://commons.wikimedia.org/wiki/File:HBV_Genome.svg
Many genes produce transcripts from both strands of DNA at the same locus. Up to 70% of mammalian genes show evidence of antisense transcription. These natural antisense transcripts (NATs) can regulate the sense gene through various mechanisms https://www.nature.com/articles/nrm2738
The Xist/Tsix pair (see X chromosome inactivation section) is a well-known example — Tsix is an antisense transcript that represses Xist on the active X
Entirely distinct genes can reside within the introns of larger genes, transcribed independently and often from the opposite strand. The NF1 gene (~350 kb, 60 exons) has three genes inside its largest intron (intron 27b, ~60 kb), all on the opposite strand:
- OMGP — oligodendrocyte-myelin glycoprotein, inhibits neurite outgrowth
- EVI2B — transmembrane glycoprotein required for granulocytic differentiation
- EVI2A — putative transmembrane protein involved in hematopoietic specification
None carry mutations causative for neurofibromatosis — they are regulated independently of NF1.
https://pmc.ncbi.nlm.nih.gov/articles/PMC359746/
Many mRNAs contain small open reading frames in their 5' UTR, upstream of the main protein-coding ORF. These uORFs are translated by ribosomes before they reach the main start codon, and can regulate translation of the downstream protein — often by reducing its translation efficiency, but sometimes by producing small functional peptides. About 50% of human mRNAs contain uORFs, yet they are routinely ignored by gene annotation pipelines.
https://en.wikipedia.org/wiki/Upstream_open_reading_frame
Chimeric genes (also called gene fusions) are formed when parts of two or more previously separate genes combine into a single new gene. This can happen through several mechanisms: retrotransposition of an mRNA into or near another gene, tandem duplication followed by fusion, unequal crossing-over between paralogs, transposon-mediated capture of gene fragments, or even readthrough transcription followed by retroposition. While many chimeric genes are nonfunctional, some acquire novel functions and are positively selected.
The gene Jingwei is a chimera (or fusion) of two genes, alcohol dehydrogenase and yellow emperor in Drosophila. Many chimeras are damaging but this has been selected for. Remarkably, two other chimeric genes (Adh-Finnegan and Adh-Twain) also independently derived from Adh via retroposition in different Drosophila lineages -- parallel chimeric gene evolution from the same parent gene http://www.pnas.org/content/101/46/16246 https://www.pnas.org/doi/10.1073/pnas.0503528102
Two Cytochrome P450 genes that don't confer any insecticide resistance on their own but a chimeric P450 does https://pubmed.ncbi.nlm.nih.gov/22949643/
SDIC in Drosophila melanogaster is a chimera of annexin X and cytoplasmic dynein intermediate chain that converted a cytoplasmic motor protein into a sperm-tail motor protein by replacing the original functional domain. Present in ~10 tandem copies on the X chromosome, it boosts sperm competition https://www.nature.com/articles/25126
Sphinx is a chimeric gene formed by retroposition of an ATP synthase gene that accumulated nonsense mutations and became a noncoding RNA. Despite losing all coding potential, it regulates male courtship behavior in Drosophila -- knockout causes increased male-male courtship https://www.pnas.org/doi/10.1073/pnas.072066399
TRIMCyp is a fusion of TRIM5 and cyclophilin A created when L1 retrotransposition inserted CypA cDNA into TRIM5. The fusion protein restricts HIV-1 infection. The same chimeric gene arose independently in both owl monkeys and pigtailed macaques via separate retrotransposition events -- convergent evolution at the gene-structure level https://www.pnas.org/doi/10.1073/pnas.0404640101
PIPSL was created when two adjacent genes (PIP5K1A and PSMD4) were transcribed as a readthrough mRNA, reverse-transcribed by L1 retrotransposon, and inserted elsewhere in the genome. Different hominoid species are at different stages of keeping or losing this gene https://genome.cshlp.org/content/17/8/1129.full
The antifreeze glycoprotein (AFGP) gene in Antarctic notothenioid fish evolved from a trypsinogen gene. The entire protein core was replaced by de novo amplification of a 9-nucleotide repeat from a tiny element at the boundary of trypsinogen's first intron. The resulting protein has zero sequence homology to its parent gene https://www.pnas.org/doi/10.1073/pnas.94.8.3811
Transposons can capture fragments of host genes during excision or replication and carry them to new genomic locations, creating chimeric genes. This "transduplication" occurs across multiple transposon superfamilies and is a major source of new genes, especially in plants.
Pack-MULEs in rice carry fragments from >1,000 genes, with ~23% fusing fragments from multiple loci into chimeric ORFs. Also found in maize and the dicot Lotus japonicus, suggesting this predates the monocot-dicot split https://www.nature.com/articles/nature02953
Helitrons (rolling-circle transposons) are the main gene-capturing transposons in animals, found in maize, bats, fungi, insects, and nematodes https://www.nature.com/articles/ncomms10716
Gene capture has been observed in real time with Pack-CACTA elements in Arabidopsis https://academic.oup.com/nar/article/47/3/1311/5198529
A broad survey found gene-capturing "Pack-TYPE" elements across all major DNA transposon superfamilies (MULEs, CACTAs, Harbingers, hATs) https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1010078
"About 70% of C. elegans mRNAs are trans-spliced to one of two 22 nucleotide spliced leaders. SL1 is used to trim off the 5' ends of pre-mRNAs and replace them with the SL1 sequence. This processing event is very closely related to cis-splicing, or intron removal."
The region that is spliced out is called an outron
http://www.wormbook.org/chapters/www_transsplicingoperons/transsplicingoperons.html

Figure from Lasda & Bhatt (2013) https://pmc.ncbi.nlm.nih.gov/articles/PMC3795323/
The leader sequence only affects the untranslated region of the mRNA, so it does not change the resulting protein.
This mechanism has evolved independently 10+ times across very different organisms. In trypanosomes, 100% of mRNAs get a spliced leader added — it is the only way to produce individual mRNAs from their polycistronic transcripts https://royalsocietypublishing.org/doi/10.1098/rsob.190072
In dinoflagellates, all mRNAs also get a spliced leader https://www.pnas.org/doi/10.1073/pnas.0700258104
The tunicate Oikopleura dioica is the only chordate known to use it https://pmc.ncbi.nlm.nih.gov/articles/PMC507004/
Copepod crustaceans use it too (discovered 2015), even though insects and most other arthropods do not https://www.nature.com/articles/srep17411
Also found in flatworms, rotifers, ctenophores, glass sponges, chaetognaths, euglenids, and cryptomonads https://academic.oup.com/mbe/article/27/3/684/1002183
The leader sequences are very similar within a phylum but totally different between phyla, suggesting many independent origins https://www.frontiersin.org/journals/genetics/articles/10.3389/fgene.2013.00199/full
The basal nematode Trichinella spiralis is an outlier — instead of the standard SL1, it has at least 15 diverse non-canonical spliced leaders https://pmc.ncbi.nlm.nih.gov/articles/PMC2271357/
Although prevalent in bacteria, operons are not common in eukaryotes. However, they are common in C. elegans specifically. "A characteristic feature of the worm genome is the existence of genes organized into operons. These polycistronic gene clusters contain two or more closely spaced genes, which are oriented in a head-to-tail direction. They are transcribed as a single polycistronic mRNA and separated into individual mRNAs by the process of trans-splicing"
http://www.wormbook.org/chapters/www_overviewgenestructure.2/genestructure.html
Another paper says "Once considered rare in eukaryotes, polycistronic mRNA expression has been identified in kinetoplastids and, more recently, green algae, red algae, and certain fungi. This study provides comprehensive evidence supporting the existence of polycistronic mRNA expression in the apicomplexan parasite Cryptosporidium parvum"
https://www.biorxiv.org/content/10.1101/2025.01.17.633476v1.full.pdf
A pre-mRNA from both strands of DNA eri6 and eri7 are combined to create eri-6/7
https://pmc.ncbi.nlm.nih.gov/articles/PMC2756026/
An example from drosophila, C. elegans, and rat shows a gene with a 5' exon being shared between two genes

https://www.fasebj.org/doi/full/10.1096/fj.00-0313rev
An example here shows 5'UTR exons shared across different olfactory receptor genes ("Some OR genes share 5'UTR exons")
https://www.biorxiv.org/content/biorxiv/early/2019/09/19/774612.full.pdf
A possible horizontal gene transfer from bacteria to eukaryotes is found in an insect that feeds on coffee beans. Changes that the gene had to undergo are covered (added poly-A tail, Shine-Dalgarno sequence deleted)
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3306691/
also https://www.cell.com/cell/fulltext/S0092-8674(19)30097-2
"The most common start codons for known Escherichia coli genes are AUG (83% of genes), GUG (14%) and UUG (3%)"
"Here, we systematically quantified translation initiation of green fluorescent protein (GFP) from all 64 codons and nanoluciferase from 12 codons on plasmids designed to interrogate a range of translation initiation conditions."
https://www.sciencedaily.com/releases/2017/02/170221080506.htm
Testing in eukaryotes has also revealed alternative starts being viable https://en.wikipedia.org/wiki/Start_codon#Eukaryotes
3-base codon system is assumed by many, but engineered tRNAs can decode 4-base codons with potential applications for using amino acids outside the 20 canonical ones
review https://elifesciences.org/articles/78869
evolving improved 4-base efficiency https://www.nature.com/articles/s41467-021-25948-y
The standard DNA double stranded helix is called B-DNA
"There are also triple-stranded DNA forms and quadruplex forms such as the G-quadruplex and the i-motif. " https://en.wikipedia.org/wiki/Nucleic_acid_double_helix
Seen in trypanosomes. The kinetoplast DNA network contains two types of circles: maxicircles (~20-40 kb) encode mitochondrial genes, but many of their transcripts are "cryptogenes" — essentially unreadable without extensive RNA editing. The guide RNAs that direct this editing are encoded on thousands of minicircles (~1 kb each) in the same network. See also the RNA editing section.

Figure from https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/kinetoplast-dna
https://pmc.ncbi.nlm.nih.gov/articles/PMC4835692/
Mitochondrial genomes are usually a single circular (or linear) molecule, but in dinoflagellates they are broken into multiple small linear chromosomes (~6-10 kb each). Only 3 protein-coding genes remain (cox1, cox3, cob), and even those are split into fragments that must be separately transcribed and pieced together https://academic.oup.com/mbe/article/24/7/1528/987488
https://en.wikipedia.org/wiki/Triple-stranded_DNA
Some organisms, famously insects in their salivary glands, create many copies of genes through multiple phases of incomplete DNA replication https://en.wikipedia.org/wiki/Polytene_chromosome

Figure source https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5768140/
"Polytene chromosomes are produced by endoreplication, in which chromosomal DNA undergoes repeated replication without cell division. Ten rounds of endoreplication produces 2^10 = 1,024 DNA strands, which when arranged alongside of each other produce distinctive banding patterns. Endoreplication occurs in cells of the larval salivary glands of many species of Diptera, and increases production of mRNA for Glue Protein that the larvae use to anchor themselves to the walls of (for example) culture vials." from https://www.mun.ca/biology/scarr/Polytene_Chromosomes.html
The above section about polytene chromosomes mentions endoreplication but this can also affect many other contexts and was mentioned as an issue in genome assembly of some plants. A talk given about vanilla bean found a lot of endoreplication during their genome assembly which leads to very uneven coverage. They tried to select tissue samples that had the least amount of endoreplication. https://plan.core-apps.com/pag_2023/abstract/e26dbeb1-df8f-4c57-a062-dcaf881b79f4
Different cells may have different numbers of copies of chromosomes and it also occurs in some human cell types: "polyploid cells can exist in otherwise diploid organisms (endopolyploidy). In humans, polyploid cells are found in critical tissues, such as liver and placenta. A general term often used to describe the generation of polyploid cells is endoreplication, which refers to multiple genome duplications without intervening division/cytokinesis" https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4442802/
"While we commonly assume the genome to be largely identical across different cells of a multicellular organism, a number of species undergo a developmental regulated elimination process by which the genome in somatic cells is reduced, while the germline genome remains intact. This process, called Programmed DNA Elimination (PDE), affects a number of species including copepod crustaceans, lamprey fish, single-celled ciliates and nematode worms (though not C. elegans!)."
From ISMB2023 video "Deciphering developmentally programmed DNA elimination in Mesorhabditis nematodes" https://www.youtube.com/watch?v=2x6ElKeISRY
Ciliates are the most extreme case — spirotrich ciliates like Oxytricha can eliminate over 95% of their germline genome when building the somatic macronucleus, including virtually all transposons. Even in Tetrahymena and Paramecium, 20-30% of the germline genome is discarded https://pmc.ncbi.nlm.nih.gov/articles/PMC3839606/
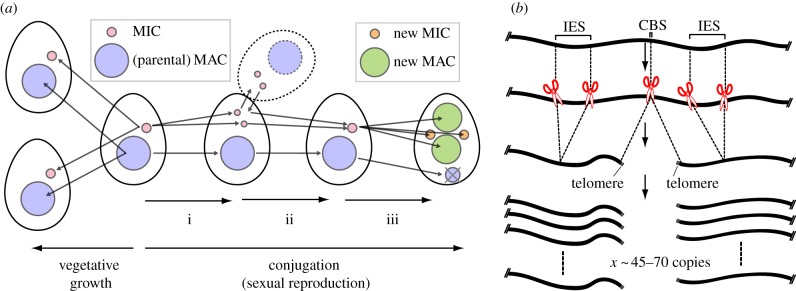
Figure from Noto & Mochizuki (2017) https://pmc.ncbi.nlm.nih.gov/articles/PMC5666084/
See also the term "internal eliminated sequences" (IES)
"How and when organisms edit their own genomes" https://www.nature.com/articles/s41588-025-02230-1
Programmed DNA elimination, VDJ recombination, CRISPR, germline restricted chromosomes, and more are examples of self-editing (quote from article: "Notable examples include the somatic diversification of immunoglobulin genes, which is the foundation of the vertebrate immune system, and natural CRISPR spacer arrays in bacteria, which recognize and cleave foreign DNA")
"Germline-restricted chromosomes (GRCs) are accessory chromosomes that occur only in germ cells. They are eliminated from somatic cells through programmed DNA elimination during embryo development. GRCs have been observed in several unrelated animal taxa and show peculiar modes of non-Mendelian inheritance and within-individual elimination. "
https://pmc.ncbi.nlm.nih.gov/articles/PMC9508068/
Some chromosomes "cheat" — they are preferentially passed to certain daughter cells instead of the expected 50/50 split, letting them spread faster than Mendelian genetics predicts. Related to meiotic drive and gene drives.
https://en.wikipedia.org/wiki/Non-random_segregation_of_chromosomes
See https://github.com/cmdcolin/oddbiology/ for more examples and details
Wikipedia lists this table with examples of organisms with different ploidy https://en.wikipedia.org/wiki/Polyploidy#Types
- haploid (one set; 1x), for example male European fire ants
- diploid (two sets; 2x), for example humans
- triploid (three sets; 3x), for example sterile saffron crocus, or seedless watermelons, also common in the phylum Tardigrada[7]
- tetraploid (four sets; 4x), for example, Plains viscacha rat, Salmonidae fish,[8] the cotton Gossypium hirsutum[9]
- pentaploid (five sets; 5x), for example Kenai Birch (Betula kenaica)
- hexaploid (six sets; 6x), for example some species of wheat,[10] kiwifruit[11]
- heptaploid or septaploid (seven sets; 7x)
- octaploid or octoploid, (eight sets; 8x), for example Acipenser (genus of sturgeon fish), dahlias
- decaploid (ten sets; 10x), for example certain strawberries
- dodecaploid or duodecaploid (twelve sets; 12x), for example the plants Celosia argentea and Spartina anglica [12] or the amphibian Xenopus ruwenzoriensis.
- tetratetracontaploid (forty-four sets; 44x), for example black mulberry[13]
There are many chemical modifications that can happen to DNA, leading to an "extended alphabet" with functional changes.
A common DNA modification is called methylation. The most common is a 5mC modification, a methylation of the letter C, and is mostly found in a CpG (a C followed by a G in the genome)
Many other modifications exist, see https://dnamod.hoffmanlab.org/
https://www.hindawi.com/journals/jna/2011/408053/tab1/
updated link on hindawi should point here http://mods.rna.albany.edu/mods/ (this link now dead too, see maybe http://genesilico.pl/modomics/modifications)
RNA editing is a post-transcriptional modification to the mRNA, which can change what we would see when the RNA is sequenced. A-to-I editing is common in some species, which would make the RNA, when sequenced, appear to have a G instead of an A. If the genome was sequenced, it would not show a SNP but the RNA-seq would appear to have A->G.
RNA editing can be conditional; mammalian apolipoprotein B is synthesized as a full-length B100 form (550 kDa) by default, but C-to-U editing in the intestine introduces a premature stop codon to produce the shorter B48 form (265 kDa)
Other editing also occurs https://en.wikipedia.org/wiki/RNA_editing
In trypanosome mitochondria, extensive U-insertion/deletion editing can more than double the length of some transcripts — the genomic sequence is essentially a compressed template that is "decoded" by editing. The instructions for editing come from guide RNAs encoded on minicircles in the kinetoplast DNA (see maxicircle architecture section). https://en.wikipedia.org/wiki/RNA_editing#Kinetoplastids
While the exon structure of most mRNAs follows the linear sequence of the transcribed DNA, there are a few cases where mature mRNAs contain exons in a non-linear order.
Al-Balool and Weber et al (2011) validated several cases of PTES in human genes that are evolutionarily conserved, including MAN1A2, PHC3, TLE4, and CDK13: https://genome.cshlp.org/content/21/11/1788.short
Maternal RNAs can show activity in the zygote (e.g. https://en.wikipedia.org/wiki/Maternal_to_zygotic_transition), which can lead to complex transgenerational effects
A lncRNA VELUCT almost flies under the radar in a lung cancer screen due to being very lowly expressed, such that it is "below the detection limit in total RNA from NCI-H460 cells by RT-qPCR as well as RNA-Seq", however this study confirms it as a factor in experiments (2025 note: this article has basically no citations, unclear whether it actually is impactful)
https://www.ncbi.nlm.nih.gov/pubmed/28160600?dopt=Abstract
Note that X inactivation relies on relatively lowly expressed RNA also https://twitter.com/mitchguttman/status/1454256452990734336
X inactivation is a form of dosage compensation — equalizing X-linked gene expression between XX and XY individuals. In mammals, one X in each female cell is silenced early in development by Xist, a long non-coding RNA that spreads along the chromosome in cis, recruiting silencing complexes. An antisense transcript Tsix represses Xist on the active X.
Not all genes on the inactive X are silenced — about 15-25% of human X-linked genes "escape" inactivation and are expressed from both copies. This is relevant to bioinformatics because these escapee genes show biallelic expression in females where you might expect monoallelic.
https://en.wikipedia.org/wiki/X-inactivation
https://www.youtube.com/watch?v=y3ST0whbA4k (great series from iBiology on X chromosome inactivation)
See https://github.com/cmdcolin/oddbiology/ for calico cats, mosaicism, and how different organisms solve dosage compensation in completely different ways
There are many types of RNA some more weird and exotic than others, a large list https://en.wikipedia.org/wiki/List_of_RNAs
Some are named based on where they are expressed or active
Others are uniquely shaped. There are also circular RNA for example https://en.wikipedia.org/wiki/Circular_RNA
Small and long non coding RNAs often fold into important structural shapes
This is probably obvious to many people who work on proteins but while the genome has almost all genes starting with a start codon which produces methionine, this is often post translationally removed https://en.m.wikipedia.org/wiki/Methionyl_aminopeptidase
An intein is a segment of a protein that catalyzes its own excision after translation, joining the flanking segments (exteins) together. Like introns for RNA, but at the protein level. https://en.wikipedia.org/wiki/Intein
See section here https://github.com/The-Sequence-Ontology/Specifications/blob/master/gff3.md#pathological-cases
Viral sequences can create a polyprotein which is fully transcribed and translated before being cleaved by a protease. In some viruses (such as coronaviruses) their translation involves ribosomal frameshifting.
Dengue, HIV, etc. use this
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6040172/ https://www.sciencedirect.com/science/article/abs/pii/S0959440X15000597
Some viruses use an alternative mechanism: 2A peptides cause the ribosome to skip forming a peptide bond during translation, effectively splitting the polyprotein without a protease. These are now widely used as a biotechnology tool for expressing multiple proteins from one mRNA https://pmc.ncbi.nlm.nih.gov/articles/PMC5438344/
From another repo https://github.com/molstar/molstar/blob/master/docs/docs/misc/interesting-pdb-entries.md
Transposable elements make up ~45% of the human genome (https://www.nature.com/articles/nature01262) and up to 85% of some plant genomes like maize (https://www.science.org/doi/10.1126/science.1178534).
Or "How a quarter of the cow genome came from snakes" http://phenomena.nationalgeographic.com/2013/01/01/how-a-quarter-of-the-cow-genome-came-from-snakes/
Source http://www.pnas.org/content/110/3/1012.full
LINE1 elements are normally kept silenced by the host, but evidence shows that LINE1 transcription is important for embryonic development.
https://www.ucsf.edu/news/2018/06/410781/not-junk-jumping-gene-critical-early-embryo
The RAG1/RAG2 recombinase that performs V(D)J recombination (see Immunity) derives from an ancient Transib transposon. The vertebrate adaptive immune system is essentially a domesticated transposon.
https://www.nature.com/articles/nature03564
CENPB, which binds centromeric DNA, derives from a pogo-like transposase https://www.nature.com/articles/384567a0
See also https://github.com/cmdcolin/oddbiology/ for syncytins and Arc (retroviral elements co-opted for placental development and neuronal communication)
Replicate via rolling-circle mechanism, can capture and shuffle host gene fragments to create chimeric genes. ~2% of the maize genome.
https://en.wikipedia.org/wiki/Helitron_(biology)
Giant DNA transposons (~15-20 kb) encoding their own DNA polymerase. May be evolutionary ancestors of some DNA viruses.
https://en.wikipedia.org/wiki/Polinton
VDJ recombination is a process of somatic DNA recombination in immune cells.
In bio 101 you learn: a gene is made of exons separated by introns, the cell transcribes the gene into RNA, introns are spliced out, and the exons are joined together in the mRNA. The DNA itself is never changed — every cell in your body has the same genome.
VDJ recombination breaks this rule. The genome contains arrays of V (variable), D (diversity), and J (joining) gene segments — many copies of each type, arranged in tandem. Instead of just transcribing and splicing RNA, immune cells physically cut the chromosomal DNA, join one V to one D to one J segment, and delete the intervening DNA. Which segments end up joined is essentially random. This is a permanent, irreversible change to the genome of that cell. The result is a brand-new exon that didn't exist in the germline genome. Only then does normal RNA splicing happen to join this newly assembled VDJ exon to the constant region exons downstream — as Janeway's Immunobiology puts it: "To make a complete immunoglobulin light-chain messenger RNA, the V-region exon is joined to the C-region sequence by RNA splicing after transcription."

Figure from https://commons.wikimedia.org/wiki/File:VDJ_recombination.png
This is why V, D, and J are called "gene segments" rather than "exons" — they are raw parts that must be assembled at the DNA level first, before the normal exon-intron splicing machinery can do its job.
The recombination is guided by flanking "recombination signal sequences" and is deliberately imprecise — terminal transferase adds random nucleotides at the junctions, further diversifying the sequences. This is how a limited number of gene segments (~100 V + ~30 D + ~6 J in humans) can generate billions of different antibodies.
This is called "somatic" recombination because it happens in somatic cells (developing B and T cells), not in the germline — the rearrangements are not passed to offspring. In most biology, recombination only occurs during meiosis in germ cells, where homologous chromosomes exchange segments. VDJ recombination is fundamentally different: it uses a dedicated enzyme complex (RAG1/RAG2) that evolved from an ancient transposon (see Transib above). RAG cuts at specific recombination signal sequences, and the broken ends are repaired by the cell's general-purpose NHEJ (non-homologous end joining) DNA repair pathway. So VDJ recombination is possible in somatic tissue because the cell already has the DNA repair machinery — RAG just co-opts it by making targeted double-strand breaks at the right places. Most somatic cells do not express RAG, which is why only lymphocytes undergo this rearrangement.
https://en.wikipedia.org/wiki/V(D)J_recombination https://www.ncbi.nlm.nih.gov/books/NBK27140/
The MHC region is a very polymorphic region of the genome on chr6. I'm not personally familiar with all the intricacies of MHC beyond that it is a unique contributor of some additional hg38 alternative loci/contigs due to its high diversity
In the Maize genome, Mo17, sequenced "T2T", they mention this very large simple repeat in the abstract...cause it's that silly
https://www.nature.com/articles/s41588-023-01419-6
A tandem duplication can be seen as a piece of DNA that copied side by side in the genome. But why would this occur?
Some biological factors can include
- replication slippage
- retrotransposition
- unequal crossing over (UCO).
- imperfect repair of double-strand breaks by nonhomologous end joining (NHEJ) (specifically generates 1-100bp range indels according to article)
Ref https://academic.oup.com/mbe/article/24/5/1190/1038942
Segmental duplications (SDs) are blocks of DNA (1 kb to hundreds of kb) present in two or more copies with >90% sequence identity. About 5-7% of the human genome is segmentally duplicated. They are a major headache for genome assembly — high identity causes collapsed or missing regions. T2T-CHM13 increased known SD content from 167 Mbp (5.4%) to 218 Mbp (7.0%), accounting for nearly one-third of newly assembled sequence.
https://www.science.org/doi/10.1126/science.abj6965
SDs serve as substrates for generating copy number variants (CNVs) via non-allelic homologous recombination (NAHR): meiotic recombination between paralogous copies instead of true alleles produces deletions, duplications, or inversions. Other CNV mechanisms include NHEJ, FoSTeS/MMBIR (replication-based template switching), and BFB cycles.
https://www.nature.com/articles/nrg2593
Medically important examples of NAHR between flanking SDs:
- 22q11.2 deletion (DiGeorge) — most common human microdeletion (~1:3,000) https://pmc.ncbi.nlm.nih.gov/articles/PMC1310636/
- CMT1A / HNPP at 17p12 — 1.5 Mb duplication (CMT1A) or reciprocal deletion (HNPP) affecting PMP22 dosage https://omim.org/entry/118220
- SMA — SMN1 loss in one of the most complex SD regions; most assemblies still don't correctly resolve SMN1/SMN2 https://pubmed.ncbi.nlm.nih.gov/15470363/
- Prader-Willi / Angelman at 15q11-q13 — same deletion, different syndrome depending on parent-of-origin (imprinting) https://pmc.ncbi.nlm.nih.gov/articles/PMC4449422/
Cancer genomes harbor catastrophic rearrangements beyond simple deletions and duplications:
- Chromothripsis — chromosome shattering and random reassembly in a single event, in >50% of some cancer types https://www.nature.com/articles/s41588-019-0576-7
- Chromoplexy — chains of balanced translocations across up to eight chromosomes, prevalent in prostate cancer (>60%) https://pmc.ncbi.nlm.nih.gov/articles/PMC3673705/
- Chromoanasynthesis — replication-based template switching producing localized duplications/triplications https://www.nature.com/articles/nm.2988
- Extrachromosomal DNA (ecDNA) — circular DNA (~1-5 Mb) replicating autonomously, driving oncogene amplification (EGFR, MYC) in ~17% of cancers. Lacks centromeres so segregates unequally https://www.nature.com/articles/s41576-022-00521-5
- Breakage-fusion-bridge (BFB) cycles — telomere loss causes sister chromatid fusion, anaphase bridge, and breakage, repeating to produce staircase amplification. Can trigger chromothripsis and ecDNA https://www.nature.com/articles/s41467-023-41259-w
- Kataegis — localized hypermutation clusters caused by APOBEC deaminases on single-stranded DNA at break sites https://elifesciences.org/articles/00534
- Tyfonas, Pyrgo, Rigma — novel complex SV classes: tyfonas (fold-back inversions, acral melanoma), pyrgo (tandem duplications, breast/ovarian), rigma (deletions at fragile sites, GI cancers) https://www.cell.com/cell/fulltext/S0092-8674(20)30997-1
The LIF gene has many copies in Elephant but many are non-functional. One copy can be "turned back on" and play a role in cancer protection. They call this a "zombie gene"
https://www.cell.com/cell-reports/fulltext/S2211-1247(18)31145-8
https://www.sciencealert.com/lif6-pseudogene-elephant-tumour-suppression-solution-petos-paradox
It has been shown that some intron sequences can enhance expression similar to how promoter sequences work https://en.wikipedia.org/wiki/Intron-mediated_enhancement
The first intron of the UBQ10 gene in Arabidopsis exhibits IME, and "the sequences responsible for increasing mRNA accumulation are redundant and dispersed throughout the UBQ10 intron" http://www.plantcell.org/content/early/2017/04/03/tpc.17.00020.full.pdf+html
The classic peppered moth phenotype is an intron TE insertion https://www.nature.com/articles/nature17951 (may not be strictly IME, I'm personally not sure)
Imprinted genes are expressed from only one parental allele. In humans, ~100-200 genes are imprinted. Classic examples: IGF2 (paternal only) and H19 (maternal only) share a regulatory region on chr11. The same chromosomal deletion at 15q11-q13 causes Prader-Willi syndrome if inherited from dad, or Angelman syndrome if from mom.
https://en.wikipedia.org/wiki/Genomic_imprinting
Wikipedia https://en.wikipedia.org/wiki/Promoter_(genetics)#Bidirectional_(mammalian)
"Bidirectional promoters are a common feature of mammalian genomes. About 11% of human genes are bidirectionally paired."
"The two genes are often functionally related, and modification of their shared promoter region allows them to be co-regulated and thus co-expressed"
See also 'divergently paired genes' https://academic.oup.com/gpb/article/23/4/qzaf058/8174973?login=false&utm_source=etoc&utm_campaign=gpb&utm_medium=email&nbd_source=campaigner&nbd=45398946253
A child can inherit both copies of a particular chromosome from one parent, instead of the "usual" one copy from mom, one from dad
"UPD arises usually from the failure of the two members of a chromosome pair to separate properly into two daughter cells during meiosis in the parent’s germline (nondisjunction). The resulting abnormal gametes contain either two copies of a chromosome (disomic) or no copy of that chromosome (nullisomic), instead of the normal single copy of each chromosome (haploid). This leads to a conception with either three copies of one chromosome (trisomy) or a single copy of a chromosome (monosomy). If a second event occurs by either the loss of one of the extra chromosomes in a trisomy or the duplication of the single chromosome in a monosomy, the karyotypically normal cell may have a growth advantage as compared to the aneuploid cells. UPD results primarily from one of these “rescue” events"
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3111049/
Older men can have a mosaic loss of the Y chromosome https://en.wikipedia.org/wiki/Mosaic_loss_of_chromosome_Y
https://www.karger.com/Article/FullText/508564 (found from https://www.biostars.org/p/9482437/)
may be associated with cardiac issues https://www.science.org/doi/10.1126/science.abn3100
Similar to the above but for X https://www.cancer.gov/news-events/press-releases/2024/genetic-factors-predict-x-chromosome-loss
In organisms with normally linear chromosomes, circular or "ring" chromosomes can form from aberrant processes https://en.wikipedia.org/wiki/Ring_chromosome

{kind=link}
{kind=link}
There are also smaller fragments that can be circularized called "supernumerary small ring chromosomes" (sSRC) or their normal linear part, "supernumary small marker chromosomes" (sSMC) https://en.wikipedia.org/wiki/Small_supernumerary_marker_chromosome
The latest human genome, for example, downloaded from NCBI, contains a number of Non-ACGT letters in the form of IUPAC codes https://www.bioinformatics.org/sms/iupac.html These represent ambiguous bases.
Here is the incidence of non-ACGTN IUPAC letters in the entire human genome GRCh38.p14 from https://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/annotation/GRCh38_latest/refseq_identifiers/GRCh38_latest_genomic.fna.gz (same for the "analysis set" files in https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/000/001/405/GCA_000001405.15_GRCh38/seqs_for_alignment_pipelines.ucsc_ids/)
{
'B' => 2,
'K' => 8,
'Y' => 36,
'M' => 8,
'R' => 29,
'W' => 15,
'S' => 5
};
Did you expect that in your bioinformatics software? Note that the mouse genome (GRCm38.p5) as far as I could tell does not contain any non-ACGT IUPAC letters
See count_fasta_letters.pl for a script to count this. The UCSC hg38.fa.gz does not have any non-ACGTN letters.
Due to how dbSNP is created (based on alignments), an rs ID can occur in multiple places on the genome https://www.biostars.org/p/2323/
In response to hg38 including a colon in sequence names, which conflicts with commonly used representation of a range as chr1:1-100 for example (note: SAMv1.pdf contains a regex to help resolve this), people analyzed meta-character frequencies in sequence names samtools/hts-specs#291
ENA
# 16927
* 1
, 231
- 122563947
. 521540419
/ 236951
\ 0
: 30181
; 72892
= 186611
@ 3713
| 949
Broad(?)
12 #
527 *
357 ,
1451132 -
1492749 .
86114 /
233731 :
2034 =
17 @
1735713 |
Reference sequences
# 203
% 203
* 525
+ 1
, 496
- 154226
. 1826561
: 1577
= 26
_ 4961932
| 1098333
Note that commas in FASTA names is being suggested as an illegal character because of the supplementary alignment tag in SAM/BAM using comma separated values
Genomes such as wheat have large chromosomes averaging 806Mbp but the BAI/TBI file formats are limited to 2^29-1 ~ 536Mbp in size (this is due to the binning strategy, the max bin size is listed as 2^29). The CSI index format was created to help index BAM and tabix files with large chromosomes.
Bonus: I made a web tool to help visualize BAI files to show how the binning index works https://cmdcolin.github.io/bam_index_visualizer/
The axolotl genome has individual chromosomes that are of size 3.14 Gbp https://genome.cshlp.org/content/29/2/317.long (2019) which is almost as big as the entire human genome
The BAM and CRAM formats can only store 2^31-1 (~2.14Gbp) length chromosomes however so bgzip/tabix SAM is used (discussion samtools/hts-specs#655)
Just some honorable mentions for largest genome
- Polychaos dubium/Amoeba dubium/Chaos chaos - ~600-1300Gbp (unsequenced, 1968 back of envelope measurement, needs confirmation) https://en.wikipedia.org/wiki/Polychaos_dubium (another ref https://bionumbers.hms.harvard.edu/bionumber.aspx?&id=117342)
- Dinoflagellates - up to 250Gbp (unsequenced, 1987 book referenced in this paper, needs confirmation, has weird chromosome "rod-like" structures) https://www.nature.com/articles/s41588-021-00841-y
- Tmesipteris oblanceolata (fork fern) - ~160Gb (unsequenced) https://www.nature.com/articles/d41586-024-01567-7
- Paris japonica (canopy plant) - ~149Gbp (unsequenced) https://en.wikipedia.org/wiki/Paris_japonica
- Tmesipteris_obliqua (fern) - ~147Gbp (unsequenced) - https://en.wikipedia.org/wiki/Tmesipteris_obliqua
- South American lungfishes (Lepidosiren paradoxa) - ~91Gbp (sequenced) https://www.nature.com/articles/s41586-024-07830-1
- European mistletoe - ~90Gbp (sequenced) https://www.darwintreeoflife.org/news_item/2022-the-year-we-built-the-biggest-genome-in-britain-and-ireland/
- Antarctic krill - ~48Gbp (sequenced) https://www.cell.com/cell/pdf/S0092-8674(23)00107-1.pdf
- Neoceratodus forsteri (Australian lungfish) - ~43Gbp (sequenced) https://www.smithsonianmag.com/smart-news/australian-lungfish-has-biggest-genome-ever-sequenced-180976837/ https://www.ncbi.nlm.nih.gov/genome/?term=Neoceratodus+forsteri
- Ambystoma mexicanum (axolotl) - ~32Gbp (sequenced) https://en.wikipedia.org/wiki/Axolotl https://www.ncbi.nlm.nih.gov/genome/?term=axolotl
- Allium ursinum (wild garlic) - ~30gb https://en.wikipedia.org/wiki/Onion_Test
- Coastal redwood - ~26Gbp (sequenced) https://www.ucdavis.edu/climate/news/coast-redwood-and-sequoia-genome-sequences-completed https://www.ncbi.nlm.nih.gov/genome/?term=redwood
- Loblolly pine - ~22Gbp (sequenced) https://blogs.biomedcentral.com/on-biology/wp-content/uploads/sites/5/2014/03/genomelog030.jpg https://www.ncbi.nlm.nih.gov/genome/?term=loblolly+pine
- Wheat genome - ~17Gbp https://academic.oup.com/gigascience/article/6/11/gix097/4561661 https://www.ncbi.nlm.nih.gov/genome/?term=wheat
{kind=link}
Inspired by twitter thread https://twitter.com/PetrovADmitri/status/1506824610360168455
Also see http://www.genomesize.com/statistics.php?stats=entire#stats_top
See also the plant C-value database, which is a measurement you will sometimes see instead of base pair length https://cvalues.science.kew.org/ ("C-value is the amount, in picograms, of DNA contained within a haploid nucleus")
On the other end of the scale, some obligate endosymbionts have extremely reduced genomes:
- Nasuia deltocephalinicola — ~112 kb, the smallest known cellular genome https://academic.oup.com/gbe/article/5/9/1675/555845
- Carsonella ruddii — ~160 kb https://www.science.org/doi/10.1126/science.1134196
- Mycoplasma genitalium — ~580 kb, used as the basis for "minimal genome" studies https://www.science.org/doi/10.1126/science.270.5235.397
Meanwhile, some giant viruses have genomes larger than these bacteria — Pandoraviruses reach ~2.5 Mb, blurring the line between viruses and cellular life https://www.science.org/doi/10.1126/science.1239181
The CG tag was invented in order to store CIGAR strings longer than 64k operations, since n_cigar_opt is a uint16 in BAM. The CIGAR string is relevant only for BAM files, CRAM uses a different storage mechanism for CIGAR type data (e.g. the reference based compression).
I extracted all the genes from a number of model organism databases here https://cmdcolin.github.io/genes/
Here are some random highlights from earlier work
- Tinman - "In mutant or knockout organisms, the loss of tinman results in the lack of heart formation" https://en.wikipedia.org/wiki/Tinman_gene
- Sonic hedgehog (SHH) - named after the video game character; hedgehog mutants have 'spiky' fruit fly embryos https://en.wikipedia.org/wiki/Sonic_hedgehog
- Robotnikin - antagonist of SHH, villain of the sonic hedgehog franchise - https://pmc.ncbi.nlm.nih.gov/articles/PMC2770933/
- Heart of glass (heg) - a zebrafish gene with mutant phenotype "Individual heg myocardial cells are also thinner than wild-type" https://www.ncbi.nlm.nih.gov/pubmed/14680629
- Dracula (drc) - "we isolated a mutation, dracula (drc), which manifested as a light-dependent lysis of red blood cells" https://www.ncbi.nlm.nih.gov/pubmed/10985389 (now renamed https://zfin.org/ZDB-GENE-000928-1)
- Sleeping Beauty transposon system - https://en.wikipedia.org/wiki/Sleeping_Beauty_transposon_system
- Skywalker (sky) - https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=35359
- TIME FOR COFFEE (TIC) - "We characterize the time for coffee (tic) mutant that disrupts circadian gating, photoperiodism, and multiple circadian rhythms, with differential effects among rhythms" https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=821807
- WTF - "Some alleles of the wtf gene family can increase their chances of spreading by using poisons to kill other alleles, and antidotes to save themselves." - https://www.ebi.ac.uk/interpro/entry/IPR004982 https://www.sciencedaily.com/releases/2017/06/170620093209.htm
- Mothers against decapentaplegic - "it was found that a mutation in the gene in the mother repressed the gene decapentaplegic in the embryo. The phrase "Mothers against" was added as a humorous take-off" https://en.wikipedia.org/wiki/Mothers_against_decapentaplegic
- Saxophone (sax) - http://www.sdbonline.org/sites/fly/gene/saxophon.htm
- Beethovan (btv) - http://www.uniprot.org/uniprot/Q0E8P6
- Superman+kryptonite - https://en.wikipedia.org/wiki/Superman_(gene)
- Supervillin (SVIL) - https://www.uniprot.org/uniprot/O95425
- Wishful thinking (wit) - https://www.wikigenes.org/e/gene/e/44096.html
- Doublesex (dsx) - "The gene is expressed in both male and female flies and is subject to alternative splicing, producing the protein isoforms dsx_f in females and the longer dsx_m in males." https://en.wikipedia.org/wiki/Doublesex
- Fruitless (fru) - "Early work refers to the gene as fruity, an apparent pun on both the common name of D. melanogaster, the fruit fly, as well as a slang word for homosexual. As social attitudes towards homosexuality changed, fruity came to be regarded as offensive, or at best, not politically correct. Thus, the gene was re-dubbed fruitless, alluding to the lack of offspring produced by flies with the mutation.[10] However, despite the original name and a continuing history of misleading inferences by the popular media, fruitless mutants primarily show defects in male-female courtship, though certain mutants cause male-male or female-female courtship.[11]" https://en.wikipedia.org/wiki/Fruitless_(gene)
- Transformer (tra) - https://en.wikipedia.org/wiki/Transformer_(gene)
- Gypsy+Flamenco - https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1206375/ also described in wiki https://en.wikipedia.org/wiki/Piwi-interacting_RNA#History_and_loci
- Jockey - http://flybase.org/reports/FBgn0015952.html
- Tigger - https://www.omim.org/entry/612972
- Nanog - celtic legend https://www.sciencedaily.com/releases/2003/06/030602024530.htm (source https://twitter.com/EpgntxEinstein/status/1057359656220348417)
- Jerky (jrk) - "A deficit in the Jerky protein in mice causes recurrent seizures" https://www.genecards.org/cgi-bin/carddisp.pl?gene=JRK
- Hippo (Hpo) - https://www.wikigenes.org/e/gene/e/37247.html
- Dishevelled (Dsh) - https://en.wikipedia.org/wiki/Dishevelled
- Glass bottom boat (gbb) - "fruit fly larvae with a faulty glass bottom boat gene are transparent" https://www.thenakedscientists.com/articles/interviews/gene-month-glass-bottom-boat http://www.sdbonline.org/sites/fly/dbzhnsky/60a-1.htm
- Makes caterpillars floppy (mcf) - https://www.pnas.org/content/99/16/10742 (source https://twitter.com/JUNIUS_64/status/1081007886560608256)
- Eyeless http://flybase.org/reports/FBgn0005558.html
- Straightjaket (stj) - http://flybase.org/reports/FBgn0261041.html
- Huluwa http://science.sciencemag.org/content/362/6417/eaat1045 ref https://twitter.com/zhouwanding/status/1065960714978897921
- frameshifts or pseudogene? - check sequence - https://www.ncbi.nlm.nih.gov/gene/?term=24562233%5Buid%5D
- Bad response to refrigeration (brr) https://twitter.com/hitenmadhani/status/1149471071675924481?s=20
- Mindbomb (mib1) - https://www.sdbonline.org/sites/fly/hjmuller/mindbomb1.htm
- β'COP http://flybase.org/reports/FBgn0025724.html (https://twitter.com/DarrenObbard/status/1260613447198412800)
- King-tubby https://www.uniprot.org/uniprot/B0XFQ9 see also tubby https://www.uniprot.org/uniprot/P50586
- fucK https://www.uniprot.org/uniprot/?query=fuck&sort=score
- scarecrow https://academic.oup.com/g3journal/article/15/5/jkaf055/8071394?login=false&utm_source=etoc&utm_campaign=g3journal&utm_medium=email
- Halloween genes (spook, disembodied, phantom, shadow, shade) https://en.wikipedia.org/wiki/Halloween_genes
- VANDAL21 https://www.arabidopsis.org/servlets/TairObject?type=transposon_family&id=139
- HotDog domain - superfamily of genes/proteins https://www.wikidata.org/wiki/Q24785143 https://www.ebi.ac.uk/interpro/entry/IPR029069
- Flower/fwe - https://flybase.org/reports/FBgn0261722.html
- Brahma https://www.sdbonline.org/sites/fly/polycomb/brahma.htm
- Pokemon gene - "The Pokémon Company threatened MSKCC with legal action in December 2005 for creating an association between cancer and the media franchise, and as a consequence MSKCC is now referring to it by its gene name Zbtb7" - Pokemon/pikachu/zubat (story https://bsky.app/profile/c0nc0rdance.bsky.social/post/3k6w3gwtell2j)
- Bring lots of money (blom7α) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2781463/ https://www.uniprot.org/uniprotkb/Q7Z7F0/entry
- MAGOH - Drosophila flies produce unfit progeny when they have mutations in their mago nashi (Japanese: 孫なし, Hepburn: mago nashi, lit. 'grandchildless') gene. The progeny have defects in germplasm assembly and germline development https://www.uniprot.org/uniprotkb/P61326/entry
- IGL@ - a locus containing many immunoglobulin genes, but why the @ sign? https://en.wikipedia.org/wiki/IGL@
- Spooky toxin - https://en.wikipedia.org/wiki/Ssm_spooky_toxin (https://twitter.com/depthsofwiki/status/1712555421918245242)
- Always early (aly) - http://flybase.org/reports/FBgn0004372.html
- Lonely guy (LOG) - https://onlinelibrary.wiley.com/doi/full/10.1111/pbi.13783
- PKZILLA (very large gene) - https://www-science-org.libproxy.berkeley.edu/doi/10.1126/science.ado3290
- Dachshund (dac) "plays a role in leg development" (in flies) https://en.wikipedia.org/wiki/Dachshund_(gene)
- Blanks ("Loss of Blanks causes complete male sterility") https://www.pnas.org/doi/10.1073/pnas.1009781108
- LUMP (and with a p-element insertion p-lump) https://pmc.ncbi.nlm.nih.gov/articles/PMC3166160/
- loquacious https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=34751
- TOPLESS https://pmc.ncbi.nlm.nih.gov/articles/PMC2643930/
- hemingway https://www.ncbi.nlm.nih.gov/gene/42207
- RAVER2 https://www.ncbi.nlm.nih.gov/gene/?term=Homo+sapiens+RAVER2
- ARSE https://www.ncbi.nlm.nih.gov/gene/100627778
- XXX https://www.ncbi.nlm.nih.gov/gene/1260943
- LAZY https://www.frontiersin.org/journals/plant-science/articles/10.3389/fpls.2020.606241/full
- BABY BOOM https://pubmed.ncbi.nlm.nih.gov/30298388/
- fiz/fezzik - "embryos and adult flies completely lacking CG9509 are unusually large - hence renaming it after the gentle giant Fezzik" (from the movie The Princess Bride) https://flybase.org/reports/FBgn0030594.htm https://www.thenakedscientists.com/articles/science-features/gene-month-fezzik
- Shavenbaby https://en.wikipedia.org/wiki/Shavenbaby see also polished rice (pri)
- DAD - defender against death https://www.ncbi.nlm.nih.gov/gene/1603
- Hang (hangover) - https://www.alliancegenome.org/gene/FB:FBgn0026575
- pickpocket (ppk) - https://flybase.org/reports/FBgn0020258.html
Sometimes it is not the gene, but the allele that is named
- Bad hair day http://www.informatics.jax.org/allele/MGI:3764934
- Samba, chacha, bossa nova http://www.informatics.jax.org/allele/MGI:3708457
- Yoda http://www.informatics.jax.org/allele/MGI:3797584
Ref https://twitter.com/hmdc_mgi/status/1242893531779391496
Great illustrations of interesting biology, including information about gene names https://twitter.com/vividbiology
Many of the stories behind fly gene nomenclature is available at https://web.archive.org/web/20110716201703/http://www.flynome.com/cgi-bin/search?source=browse including the famous ForRentApartments dot com gene (just kidding but lol https://web.archive.org/web/20110716202150/http://www.flynome.com/cgi-bin/search?storyID=180)
Musing article: "What is in a (gene) name?" https://web.archive.org/web/20180731060319/https://blogs.plos.org/toothandclaw/2012/06/17/whats-in-a-gene-name/